
Discoveries in bioinformatics provide new therapeutic interventions to disease by replacing expensive, time-consuming physical experiments with an automated computational search. Public databases now contain experimentally determined sequence and structural information for hundreds of thousands of proteins allowing for rather comprehensive digital investigation. Computational software and hardware limitations are the restricting factor in obtaining deeper understanding of molecular function from this available data. Computational development of future drugs and treatments involves many tools for processing, searching, and simulating these large databases. Improvements are clearly needed to develop a core library of algorithms underpinning the computational prediction and virtual screening for scientific discovery. As available computer systems are constantly changing in various forms and factors, these algorithms must be efficiently mapped to emerging architectures to continue to tackle problems of increased realism and significance.
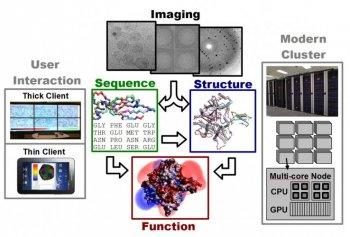
CVC research on drug discovery spans this computational drug discovery pipeline. This begins with improved tools for reconstructing accurate structural models from x-ray diffraction and electron microscopy data. With these structural models, computationally efficient biochemical/physiological models for estimating the interaction between molecules and/or chemical compounds are being enhanced to more accurately reflect the true binding affinities. These models are integrated into fast search algorithms for identifying the minimum energy binding configurations of a target with a proposed drug. Underneath these specific challenges in computational biophysics and biochemistry are more fundamental issues in computational science including the fast Fourier transform, the fast multipole method, and variational methods for solving inverse problems. To harness modern computing, these algorithms are designed to be cache aware and optimally mapped to heterogeneous mutli-core CPU and many-core GPU architectures.
By Chandrajit Bajaj, Bioinformatics and Computational Biology, Graphics and Visualization, and Scientific Computing
3D Cryo-EM | Docking | Software: VolRover | Software: TexMol